4 Simulation example
4.1 Data-generating process
The data-generating process is described via the directed acyclic graph (DAG) in Fig. 4.1. In this DAG, relative humidity \((\text{H})\) influences respiratory irritation \((\text{R})\) both directly and indirectly, passing through microbial aerosol \((\text{M})\). Additionally, respiratory irritation \((\text{R})\) influences absence \((\text{A})\) directly.
Code
dag_coords.ex4 <-
data.frame(name = c('M', 'H', 'A', 'R'),
x = c(1, 3.5, 6, 6),
y = c(3, 2, 1, 3))
DAG.ex4 <-
dagify(M ~ H,
R ~ H + M,
A ~ R,
coords = dag_coords.ex4)
node_labels <- c(
M = 'bold(M)',
H = 'bold(H)',
A = 'bold(A)',
R = 'bold(R)'
)
ggplot(data = DAG.ex4, aes(x = x, y = y, xend = xend, yend = yend)) +
geom_dag_text(aes(label = node_labels[name]),
parse = TRUE,
colour = 'black',
size = 10,
family = 'mono') +
geom_dag_edges(arrow_directed = grid::arrow(length = grid::unit(10, 'pt'), type = 'open'),
edge_colour = 'black',
family = 'mono',
fontface = 'bold') +
annotate('text', x = 1, y = 3.3, label = 'microbial aerosol',
size = 4, hjust = 0.5, colour = 'grey50') +
annotate('text', x = 3.5, y = 1.7, label = 'relative humidity',
size = 4, hjust = 0.5, colour = 'grey50') +
annotate('text', x = 6, y = 3.3, label = 'respiratory irritation',
size = 4, hjust = 0.7, colour = 'grey50') +
annotate('text', x = 6, y = 0.7, label = 'absence',
size = 4, hjust = 0.6, colour = 'grey50') +
coord_cartesian(xlim = c(0.5, 6.5), ylim = c(0.8, 3.2)) +
theme_dag()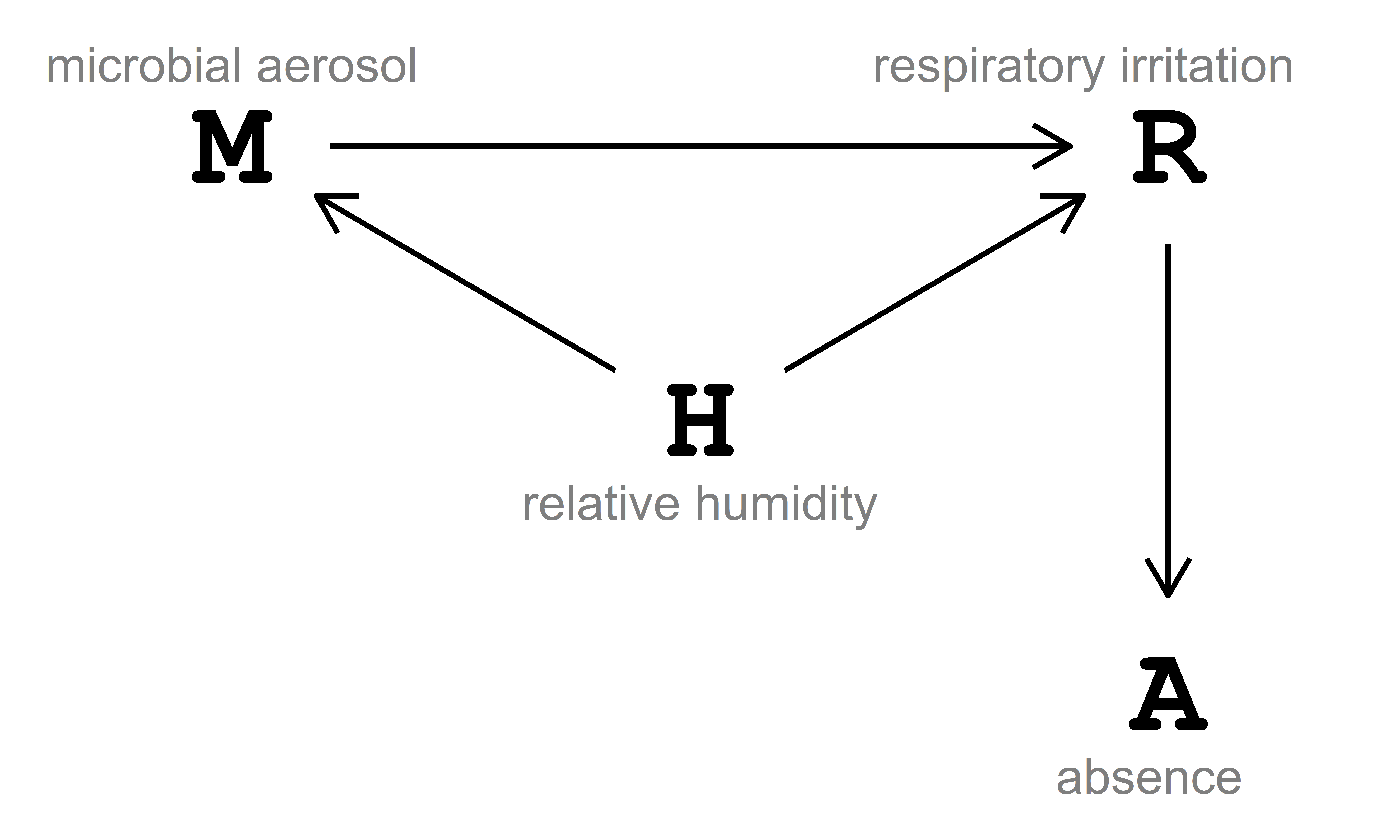
The DAG in Fig. 4.1 can be written as:
- \(M \sim f_{M}(H)\), read as ‘microbial aerosol \((\text{M})\) is some function of relative humidity \((\text{H})\)’.
- \(R \sim f_{R}(H, M)\), read as ‘respiratory irritation \((\text{R})\) is some function of relative humidity \((\text{H})\) and microbial aerosol \((\text{M})\)’.
- \(A \sim f_{A}(R)\), read as ‘absence \((\text{A})\) is some function of respiratory irritation \((\text{R})\)’.
4.2 Synthetic data set
To generate synthetic data, we defined the custom function data.sim_ex4(). This function takes as inputs the sample size n and generates synthetic data according to the DAG in Fig. 4.1.
data.sim_ex4 <- function(n) {
b_hum.resp = c(0, 0.5) #direct causal effect of H on R
b_micr.resp = c(0, 3) #direct causal effect of M on R
#Simulate relative humidity
H <- factor(sample(c('low', 'high'), size = n, replace = TRUE))
#Simulate microbial aerosol
M <- factor(ifelse(rbinom(n = n, size = 1, prob = plogis(ifelse(H == 'low', -1.5, 2))) == 0, 'low', 'high'))
#Simulate respiratory irritation
R <- rnorm(n = n, mean = -1 + ifelse(H == 'high', b_hum.resp[1], b_hum.resp[2]) + ifelse(M == 'low', b_micr.resp[1], b_micr.resp[2]), sd = 3)
#Simulate absence
A <- factor(ifelse(rbinom(n = n, size = 1, prob = plogis(0.3 + 2 * R)) == 0, 'no', 'yes'))
#Return tibble with simulated values
return(tibble(H, A, M, R))
}From this data generation mechanism, we simulated the target population, which consists of one million observations.
set.seed(2025) #set random number for reproducibility
#Simulate the population
ex4_population <- data.sim_ex4(n = 1e6)
#Set relative humidity reference category to 'high'
ex4_population$H <- relevel(ex4_population$H, ref = 'high')
#Set microbial aerosol reference category to 'low'
ex4_population$M <- relevel(ex4_population$M, ref = 'low')
#Set absence reference category to 'no'
ex4_population$A <- relevel(ex4_population$A, ref = 'no')
#View the data frame
ex4_population# A tibble: 1,000,000 × 4
H A M R
<fct> <fct> <fct> <dbl>
1 low yes low 5.95
2 high yes high 9.13
3 high yes high -0.00136
4 high no high -3.59
5 low no low -3.32
6 low yes low 0.832
7 high yes high 2.46
8 low no low -4.59
9 high yes high 6.80
10 low no low -4.98
# ℹ 999,990 more rowsFrom this population, we obtained one data set of five thousand observations using simple random sampling.
set.seed(ex4_random.seed[1])
#Sample one data set of 5,000 observations
ex4_sample.random <-
ex4_population %>%
slice_sample(n = 5e3) #take a simple random sample of size 5,000
#View the data frame
ex4_sample.random# A tibble: 5,000 × 4
H A M R
<fct> <fct> <fct> <dbl>
1 high yes high 5.94
2 low yes low 0.799
3 low no low -2.45
4 high yes high 2.25
5 low no low -1.24
6 low no low -2.90
7 high no high -0.279
8 low no low -2.21
9 high no high -2.38
10 low no low -4.50
# ℹ 4,990 more rows4.3 Data analysis
In this example, the target of our analysis is the total average causal effect, ACE (also known as total average treatment effect, ATE) of microbial aerosol \((\text{M})\) on respiratory irritation \((\text{R})\), which stands for the expected increase of \(\text{R}\) in response to a change in \(\text{M}\) from low to high due to an intervention. The causal effect of interest is visualized in Fig. 4.2.
Code
ggplot(data = DAG.ex4, aes(x = x, y = y, xend = xend, yend = yend)) +
#visualize causal effect path
geom_segment(x = 1, xend = 6, y = 3, yend = 3,
linewidth = 14, lineend = 'round', colour = '#009E73', alpha = 0.05) +
geom_dag_text(aes(label = node_labels[name]),
parse = TRUE,
colour = 'black',
size = 10,
family = 'mono') +
geom_dag_edges(arrow_directed = grid::arrow(length = grid::unit(10, 'pt'), type = 'open'),
edge_colour = 'black',
family = 'mono',
fontface = 'bold') +
annotate('text', x = 1, y = 3.3, label = 'microbial aerosol',
size = 4, hjust = 0.5, colour = 'grey50') +
annotate('text', x = 3.5, y = 1.7, label = 'relative humidity',
size = 4, hjust = 0.5, colour = 'grey50') +
annotate('text', x = 6, y = 3.3, label = 'respiratory irritation',
size = 4, hjust = 0.7, colour = 'grey50') +
annotate('text', x = 6, y = 0.7, label = 'absence',
size = 4, hjust = 0.6, colour = 'grey50') +
#causal effect number
annotate('text', x = 3.5, y = 3.2, label = '3',
size = 4.5, hjust = 0.5, colour = 'black', parse = TRUE) +
coord_cartesian(xlim = c(0.5, 6.5), ylim = c(0.8, 3.2)) +
theme_dag()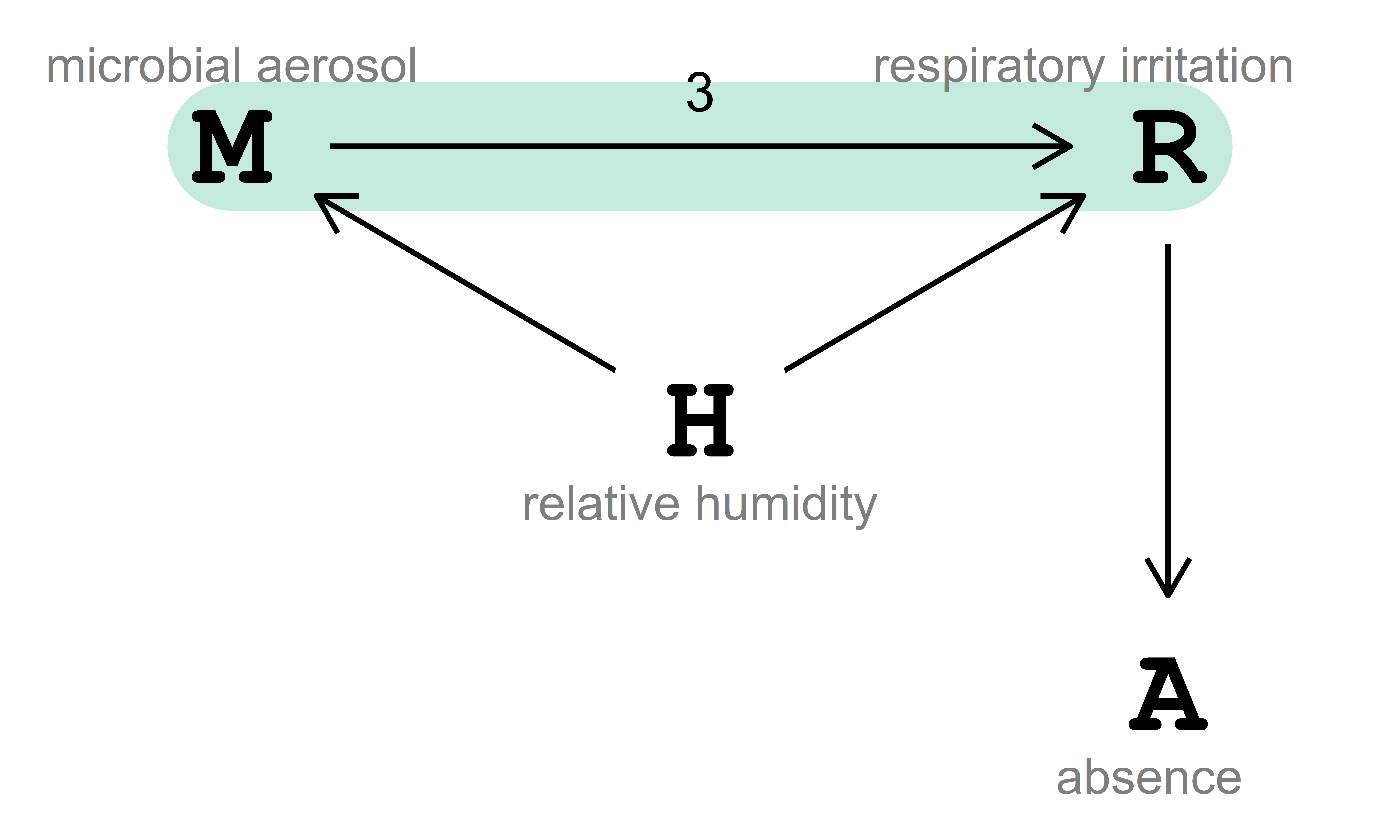
4.3.1 Identification
The first step to answer the causal question of interest is identification. Identification answers a ‘theoretical’ question by determining whether a causal effect can, in principle, be estimated from observed data. The backdoor criterion and its generalization, the adjustment criterion, allow us to understand whether our causal effect of interest can be identified and, if so, which variables we should (or should not) statistically adjust for (i.e., the adjustment set) to estimate the causal effect from the data.
Given its simplicity, we will first apply the backdoor criterion to identify valid adjustment sets to estimate the causal effect of interest. If the backdoor criterion is not applicable, we will apply its generalization, the adjustment criterion.
Backdoor criterion
Applying the adjustment criterion revealed the existence of a backdoor path (i.e., a non-causal path) from microbial aerosol \((\text{M})\) to respiratory irritation \((\text{R})\). Specifically, the backdoor path is \(\text{M} \leftarrow \text{H} \rightarrow \text{R}\). Since relative humidity \((\text{H})\) is a common cause of \(\text{M}\) and \(\text{R}\), association can flow from \(\text{M}\) to \(\text{R}\) through \(\text{H}\). As a result, there is confounding. To close this backdoor path we need to adjust for \(\text{H}\).
Given the DAG in Fig. 4.2, we can use the adjustmentSets() function to identify the adjustment set algorithmically. It is essential to note that this function applies the adjustment criterion and not the backdoor criterion. As such, the adjustmentSets() function can find adjustment sets even when the backdoor criterion is not applicable.
adjustmentSets(DAG.ex4,
exposure = 'M', #microbial aerosol
outcome = 'R', #respiratory irritation
type = 'all',
effect = 'total',
max.results = Inf){ H }As expected, the resulting adjustment set includes \(\text{H}\). Therefore, to get the correct estimate of the total average causal effect, we need to adjust for \(\text{H}\); failing to do so will lead to bias.
4.3.2 Estimation
Following the identification step is the estimation step. This step addresses a statistical question by determining how the causal effect identified in the previous step can be estimated. To perform this step, we used a parametric (model-based) estimator, specifically, linear regression. This was possible because we designed the illustrative examples to be simple and with a linear relationship between the variables. This way, we limited the complexity of the examples themselves and shifted the focus to the application of the backdoor criterion to define ‘correct’ adjustment sets.
For transparency and understanding, all (implicit) assumptions used for this illustrative example are (explicitly) provided in Table 4.1.
| Research question | Total average causal effect (ACE) of microbial aerosol (M) on respiratory irritation (R). |
| Assumptions | Random sample (simple random sampling): everyone in the population has an equal chance of being selected into the sample. |
| Limited random variability: large sample size. | |
| Independence of observations: each observation represents independent bits of information. | |
| No confounding: the DAG includes all shared causes among the variables. | |
| No model error: perfect functional form specification. | |
| No measurement error: all variables are measured perfectly. | |
| Variables | Relative humidity (H): categorical variable ['low'; 'high'] |
| Microbial aerosol (M): categorical variable ['low'; 'high'] | |
| Absence (A): categorical variable ['no'; 'yes'] | |
| Respiratory irritation (R): continuous variable [unit: -] |
To carry out the estimation step, we utilized linear regression within both the frequentist and Bayesian frameworks. Specifically, we will run three regression models:
-
Model.1will include microbial aerosol \((\text{M})\), relative humidity \((\text{H})\) and absence \((\text{A})\) as predictor; -
Model.2will include microbial aerosol \((\text{M})\) and relative humidity \((\text{H})\) as predictors.
The results of the fitted statistical models (i.e., Model.1 and Model.2) are presented here.
Frequentist framework
Click to expand
#Fit the linear regression model with M, H, A and R (Model 1)
ex4_Model.1 <-
lm(formula = R ~ M + H + A,
data = ex4_sample.random)
#View of the model summary
summary(ex4_Model.1)
Call:
lm(formula = R ~ M + H + A, data = ex4_sample.random)
Residuals:
Min 1Q Median 3Q Max
-9.725 -1.386 -0.043 1.365 7.905
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -2.95007 0.08678 -33.99 < 2e-16 ***
Mhigh 1.37308 0.08360 16.42 < 2e-16 ***
Hlow 0.32211 0.08114 3.97 7.29e-05 ***
Ayes 4.63997 0.06148 75.47 < 2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 2.013 on 4996 degrees of freedom
Multiple R-squared: 0.606, Adjusted R-squared: 0.6058
F-statistic: 2562 on 3 and 4996 DF, p-value: < 2.2e-16#Fit the linear regression model with M, H and R (Model 2)
ex4_Model.2 <-
lm(formula = R ~ M + H,
data = ex4_sample.random)
#View of the model summary
summary(ex4_Model.2)
Call:
lm(formula = R ~ M + H, data = ex4_sample.random)
Residuals:
Min 1Q Median 3Q Max
-11.8867 -1.9861 0.0083 2.0066 9.2993
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.9769 0.1210 -8.071 8.69e-16 ***
Mhigh 2.8811 0.1187 24.262 < 2e-16 ***
Hlow 0.5111 0.1186 4.309 1.67e-05 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 2.944 on 4997 degrees of freedom
Multiple R-squared: 0.1569, Adjusted R-squared: 0.1565
F-statistic: 464.9 on 2 and 4997 DF, p-value: < 2.2e-16The estimated coefficients for the two models are then plotted in Fig. 4.3.
Code
b_micr.resp = 3
data.frame(model = c('Model.1', 'Model.2'),
estimate = c(coef(ex4_Model.1)['Mhigh'],
coef(ex4_Model.2)['Mhigh']),
lower.95.CI = c(confint(ex4_Model.1, level = 0.95, type = 'Wald')['Mhigh', 1],
confint(ex4_Model.2, level = 0.95, type = 'Wald')['Mhigh', 1]),
upper.95.CI = c(confint(ex4_Model.1, level = 0.95, type = 'Wald')['Mhigh', 2],
confint(ex4_Model.2, level = 0.95, type = 'Wald')['Mhigh', 2])) %>%
ggplot(aes(x = estimate, y = model, xmin = lower.95.CI, xmax = upper.95.CI)) +
geom_vline(xintercept = b_micr.resp, alpha = 0.8, linetype = 'dashed', colour = 'blue') +
geom_linerange() +
geom_point(shape = 21, size = 2, fill = 'white', stroke = 1) +
scale_x_continuous('Estimate',
breaks = seq(from = -5, to = 5, by = 0.5),
limits = c(0.5, 3.5)) +
theme(panel.grid = element_blank(),
panel.background = element_blank(),
panel.border = element_rect(colour = 'black', fill = NA),
axis.title.y = element_blank())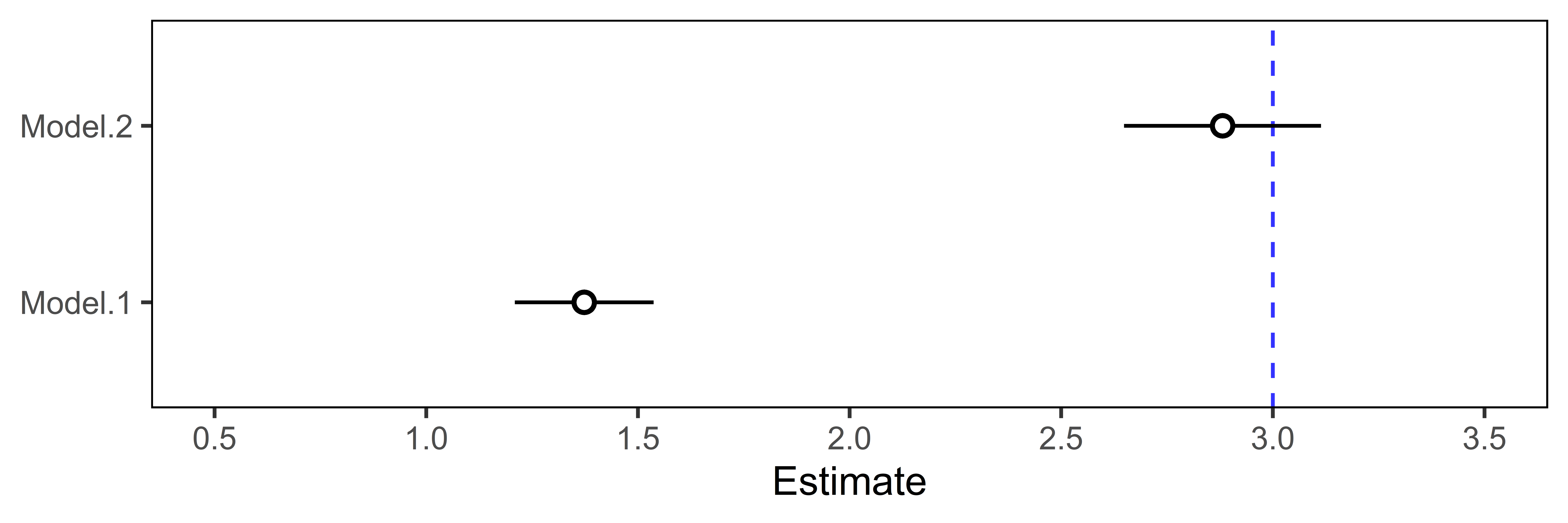
Fig. 4.3 shows the estimates (point estimate and 95% confidence interval) of the coefficient for microbial aerosol for Model.1 and Model.2.
For Model.1 we found a positive coefficient between microbial aerosol \((\text{M})\) and respiratory irritation \((\text{R})\) equal to 1.373 with 95% confidence interval (CI) [1.209, 1.537]. Since the 95% CI excludes zero, the regression coefficient is statistically significantly different from zero at the 0.05 level (p-value = 4.47e-59). Moreover, since the 95% CI does not include the data-generating parameter for microbial aerosol (i.e., b_micr.resp = 3), we can deduce that the estimated coefficient for microbial aerosol is statistically significantly different from 3 at the 0.05 level. We can test this more formally by using the linearHypothesis() function. Specifically,
#Test for M = 3
car::linearHypothesis(ex4_Model.1, 'Mhigh = 3') # car:: access the function from the car package without loading it in the environment
Linear hypothesis test:
Mhigh = 3
Model 1: restricted model
Model 2: R ~ M + H + A
Res.Df RSS Df Sum of Sq F Pr(>F)
1 4997 21775
2 4996 20240 1 1534.2 378.69 < 2.2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The resulting p-value is 2.31e-81, which indicates that we can reject the null hypothesis (i.e., M = 3) at the 0.05 level. This result suggests that the regression coefficient for microbial aerosol (i.e., 1.373) is statistically significantly different from 3. Since the estimated causal effect from Model.1 is biased, we would erroneously conclude that changing microbial aerosol from low to high causes an increase in lung infection by 1.373 units (95% CI [1.209, 1.537]).
For Model.2 we found a positive coefficient between microbial aerosol \((\text{M})\) and respiratory irritation \((\text{R})\) equal to 2.881 with 95% CI [2.648, 3.114]. Since the 95% CI excludes zero, the regression coefficient is statistically significantly different from zero at the 0.05 level (p-value = 5.01e-123). Additionally, since the 95% CI includes the data-generating parameter for microbial aerosol (i.e., b_micr.resp = 3), we can deduce that the estimated coefficient for microbial aerosol is not statistically significantly different from 3 at the 0.05 level (although this will be the case for all numbers within the 95% confidence interval). We can test this more formally by using the linearHypothesis() function. Specifically,
#Test for M = 3
car::linearHypothesis(ex4_Model.2, 'Mhigh = 3') # car:: access the function from the car package without loading it in the environment
Linear hypothesis test:
Mhigh = 3
Model 1: restricted model
Model 2: R ~ M + H
Res.Df RSS Df Sum of Sq F Pr(>F)
1 4998 43324
2 4997 43315 1 8.6974 1.0034 0.3165The resulting p-value is 0.317, which indicates that we fail to reject the null hypothesis (i.e., M = 3) at the 0.05 level. This result suggests that the regression coefficient for microbial aerosol (i.e., 2.881) is not statistically significantly different from 3. Since the estimated causal effect from Model.2 is unbiased, we would correctly conclude that changing microbial aerosol from low to high causes an increase in lung infection by 2.881 units (95% CI [2.648, 3.114]).
Importantly, for Model.1 and Model.2, the 95% confidence interval means that if we were to repeat the sampling process and calculate the interval many times, 95% of those calculated intervals would contain the true population parameter. To highlight this, we can repeat the analysis by fitting the two models to one thousand data sets randomly selected from our population.
The for-loop shown in the code below performs the following operations. First, sample (using simple random sampling) a data set of 5,000 observations from the target population. Subsequently, perform linear regression using Model.1 and Model.2 and store the estimated coefficients for microbial aerosol, its standard error and 95% confidence interval in the data frame coefs_ex4. This operation is repeated a thousand times, resulting in the data frame coefs_ex4 containing the estimates (point estimate, standard error and confidence interval) of a thousand random samples of size 5,000 using Model.1 and Model.2.
n_model <- c('mod.1', 'mod.2')
n_row <- n_sims*length(n_model)
#Create an empty data frame
empty.df <- data.frame(matrix(NA, nrow = n_row, ncol = 7))
#Rename the data frame columns
colnames(empty.df) <- c('sim.id', 'estimate', 'se', 'CI_2.5', 'CI_97.5',
'model', 'coverage')
#Sample a thousand data sets of 5,000 observations and perform linear regression
coefs_ex4 <- empty.df #assign the empty data frame
k = 1
for (i in 1:n_sims){
set.seed(ex4_random.seed[i]) #set unique seed for each simulation
#Sample data set from population
sample.random <-
ex4_population %>%
slice_sample(n = 5e3) #take a simple random sample of size 5,000
#Fit models
for (j in 1:length(n_model)){
if (n_model[j] == 'mod.1'){
fit <- lm(formula = R ~ M + H + A,
data = sample.random)
} else {
fit <- lm(formula = R ~ M + H,
data = sample.random)
}
#Compile matrix
coefs_ex4[k, 1] <- i #simulation ID
coefs_ex4[k, 2] <- coef(fit)['Mhigh'] #point estimate
coefs_ex4[k, 3] <- summary(fit)$coef['Mhigh','Std. Error'] #standard error
coefs_ex4[k, 4:5] <- confint(fit, level = 0.95, type = 'Wald')['Mhigh', ] #confidence interval (Wald)
coefs_ex4[k, 6] <- n_model[j] #sample size
k = k + 1
}
}
coefs_ex4 <- as_tibble(coefs_ex4)
#View the data frame
coefs_ex4# A tibble: 2,000 × 7
sim.id estimate se CI_2.5 CI_97.5 model coverage
<int> <dbl> <dbl> <dbl> <dbl> <chr> <lgl>
1 1 1.37 0.0836 1.21 1.54 mod.1 NA
2 1 2.88 0.119 2.65 3.11 mod.2 NA
3 2 1.30 0.0874 1.13 1.47 mod.1 NA
4 2 2.89 0.122 2.65 3.13 mod.2 NA
5 3 1.38 0.0881 1.21 1.56 mod.1 NA
6 3 3.11 0.123 2.87 3.35 mod.2 NA
7 4 1.47 0.0844 1.31 1.64 mod.1 NA
8 4 3.12 0.118 2.89 3.35 mod.2 NA
9 5 1.33 0.0841 1.17 1.50 mod.1 NA
10 5 2.99 0.118 2.76 3.22 mod.2 NA
# ℹ 1,990 more rowsThe coverage is defined by setting its value to 1 if the confidence interval overlaps the data-generating parameter for microbial aerosol (i.e., b_micr.resp = 3) and 0 otherwise.
#Calculate coverage
coefs_ex4 <-
coefs_ex4 %>%
mutate(coverage = case_when(CI_2.5 > b_micr.resp | CI_97.5 < b_micr.resp ~ 0,
CI_2.5 <= b_micr.resp & CI_97.5 >= b_micr.resp ~ 1,
.default = NA))
#View the data frame
coefs_ex4# A tibble: 2,000 × 7
sim.id estimate se CI_2.5 CI_97.5 model coverage
<int> <dbl> <dbl> <dbl> <dbl> <chr> <dbl>
1 1 1.37 0.0836 1.21 1.54 mod.1 0
2 1 2.88 0.119 2.65 3.11 mod.2 1
3 2 1.30 0.0874 1.13 1.47 mod.1 0
4 2 2.89 0.122 2.65 3.13 mod.2 1
5 3 1.38 0.0881 1.21 1.56 mod.1 0
6 3 3.11 0.123 2.87 3.35 mod.2 1
7 4 1.47 0.0844 1.31 1.64 mod.1 0
8 4 3.12 0.118 2.89 3.35 mod.2 1
9 5 1.33 0.0841 1.17 1.50 mod.1 0
10 5 2.99 0.118 2.76 3.22 mod.2 1
# ℹ 1,990 more rowsThe results are then plotted in Fig. 4.4.
Code
model_names <- c('mod.1' = 'Model.1',
'mod.2' = 'Model.2')
ggplot(data = subset(coefs_ex4, sim.id <= 2e2), aes(x = sim.id, y = estimate, ymin = CI_2.5, ymax = CI_97.5, colour = as.factor(coverage))) +
geom_hline(yintercept = b_micr.resp, alpha = 0.8, linetype = 'dashed', colour = 'blue') +
geom_linerange() +
geom_point(shape = 21, size = 1, fill = 'white', stroke = 0.5) +
scale_colour_manual('', values = c('black', '#FF4040'),
breaks = c('1','0')) +
scale_y_continuous('Estimate',
breaks = seq(from = -3, to = 3, by = 0.5)) +
scale_x_continuous('Simulation ID') +
facet_wrap(model~.,
labeller = labeller(model = model_names),
nrow = 3,
scales = 'fixed') +
theme(legend.position = 'none',
panel.grid = element_blank(),
panel.background = element_blank(),
panel.border = element_rect(colour = 'black', fill = NA),
axis.title.y = element_blank())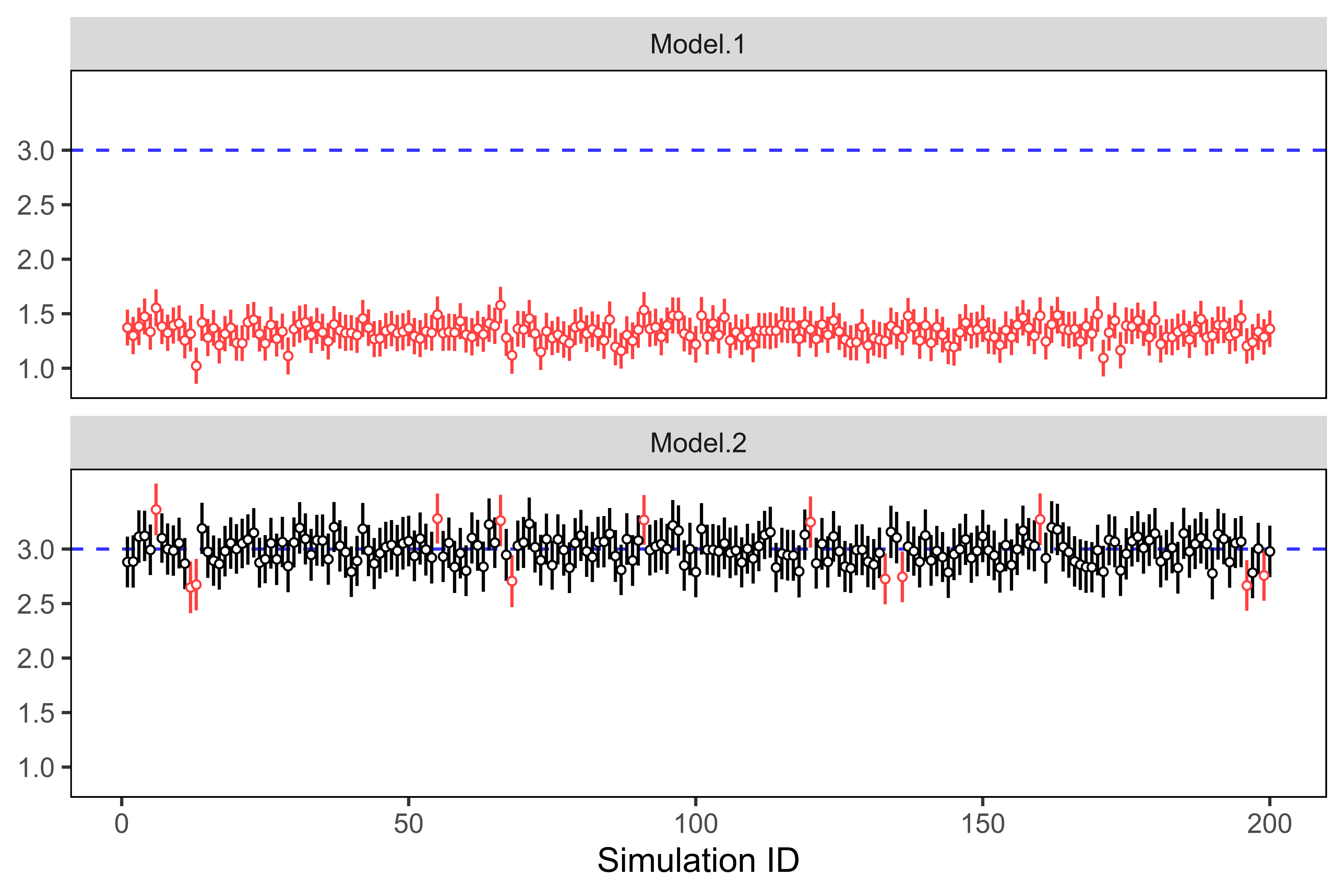
Fig. 4.4 shows the first 200 estimates (point estimate and confidence interval) of the coefficient for microbial aerosol for Model.1 and Model.2. If all the thousand simulations are considered, the frequency of the coverage of the calculated confidence intervals (i.e., how many times the confidence intervals overlap the data-generating parameter) is 0.0% and 95.3% for Model.1 and Model.2, respectively. Since the estimate of the causal effect for Model.1 are biased, the calculated confidence intervals for this model do not have the expected coverage. In fact, confidence intervals only quantify the uncertainty due to random error (i.e., sample variability), not systematic error (i.e., bias). Instead, the estimate from Model.2 are unbiased and the calculated 95% confidence intervals have the expected coverage (i.e., overlap the data-generating parameter 95% of the times).
We can also visualize all the 1,000 estimates of the coefficient for microbial aerosol for Model.1 and Model.2 (the estimates were shown as white dots in Fig. 4.4).
Code
ggplot(data = coefs_ex4, aes(x = estimate, y = ifelse(after_stat(count) > 0, after_stat(count), NA))) +
geom_histogram(binwidth = 0.05, fill = 'white', colour = 'grey50') +
geom_vline(xintercept = b_micr.resp, colour = 'blue', linetype = 'dashed') +
scale_x_continuous('Estimate',
breaks = seq(from = -5, to = 5, by = 0.5),
limits = c(0.9, 3.5)) +
facet_grid(model~.,
labeller = labeller(model = model_names),
scales = 'fixed') +
theme(axis.title.y = element_blank(),
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
panel.grid = element_blank(),
panel.background = element_blank(),
panel.border = element_rect(colour = 'black', fill = NA))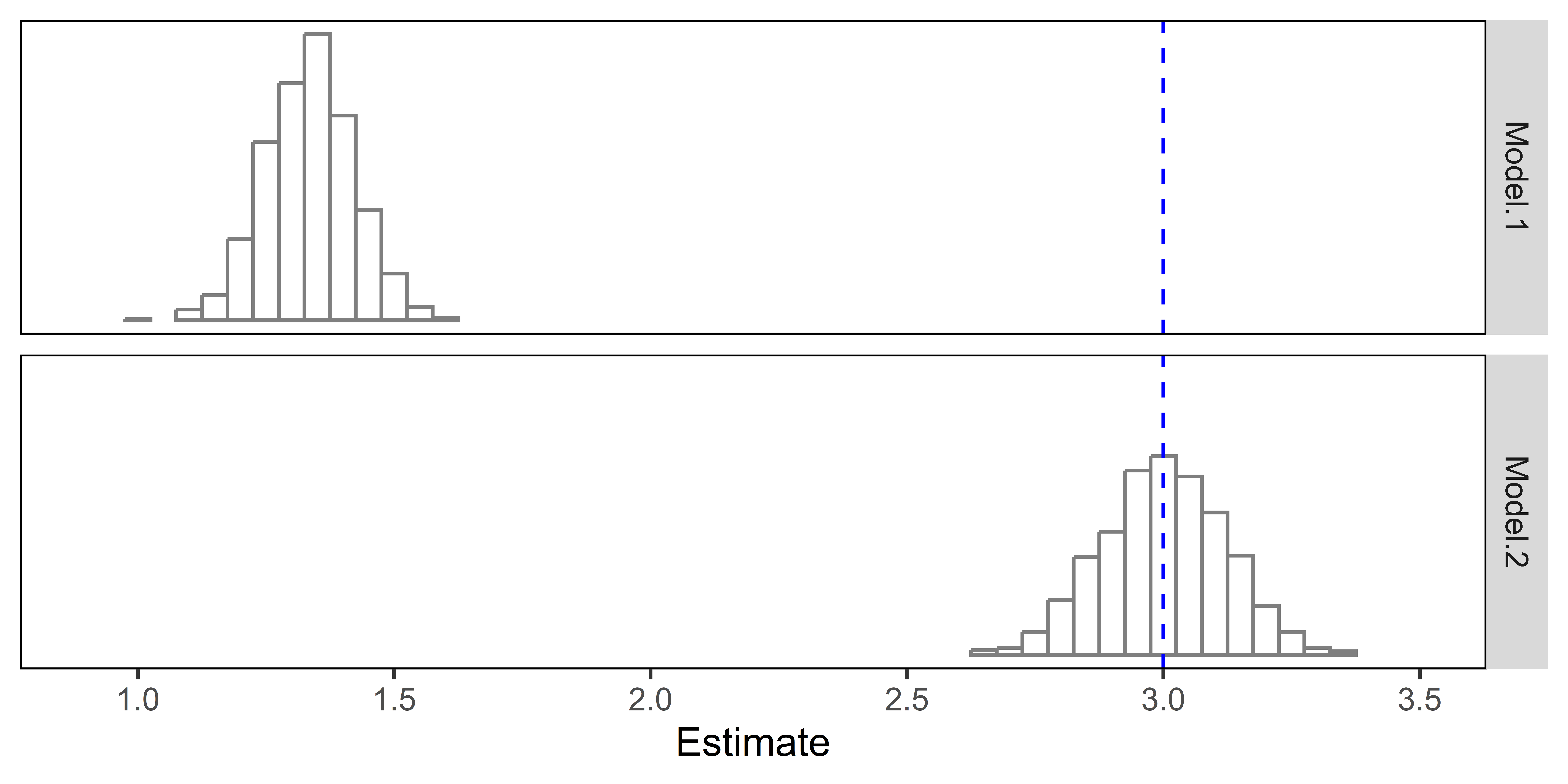
In Fig. 4.5, the estimated coefficients are visualized through a histogram. Here, we can see that the estimates for Model.1 are not clustered around the data-generating parameter (blue dashed line) having a mean estimate of 1.334 (with 0.086 standard deviation). In contrast, the estimates for Model.2 having a mean estimate of 2.999 (with 0.120 standard deviation) are centered around the data-generating parameter.
As expected, adjusting for relative humidity \((\text{H})\) (i.e., using Model.2) leads to the correct estimate of the total average causal effect of microbial aerosol \((\text{M})\) on respiratory irritation \((\text{R})\). However, when adjusting for both relative humidity \((\text{H})\) and absence \((\text{A})\) (i.e., using Model.1) the estimates are biased. This happened because absence \((\text{A})\) is a descendant of respiratory irritation \((\text{R})\), which is collider. Adjusting for a descendant of a collider is akin to adjusting for the collider itself, and, therefore, can introduce a similar bias.
To better explain this bias, we can plot the magnified DAG that shows that causes of all variables.
Code
dag_coords.ex4.3 <-
data.frame(name = c('M', 'H', 'A', 'R', 'UA', 'UH', 'UM', 'UR'),
x = c(1, 3.5, 6, 6, 8, 3.5, 1, 8),
y = c(3, 2, 1, 3, 1, 1, 2, 3))
DAG.ex4.3 <-
dagify(M ~ H + UM,
H ~ UH,
R ~ H + M + UR,
A ~ R + UA,
coords = dag_coords.ex4.3)
node_labels <- c(
M = 'bold(M)',
H = 'bold(H)',
A = 'bold(A)',
R = 'bold(R)',
UA = 'bold(UA)',
UH = 'bold(UH)',
UM = 'bold(UM)',
UR = 'bold(UR)'
)
ggplot(data = DAG.ex4.3, aes(x = x, y = y, xend = xend, yend = yend)) +
#visualize causal effect path
geom_segment(x = 1, xend = 8, y = 3, yend = 3,
linewidth = 8, lineend = 'square', colour = '#0072B2', alpha = 0.05, linetype = 'solid') +
geom_point(x = 8, y = 3, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 8, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 3.5, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 1, y = 2, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_dag_text(aes(label = node_labels[name]),
parse = TRUE,
colour = 'black',
size = 10,
family = 'mono') +
geom_dag_edges(arrow_directed = grid::arrow(length = grid::unit(10, 'pt'), type = 'open'),
edge_colour = 'black',
family = 'mono',
fontface = 'bold') +
coord_cartesian(xlim = c(0.5, 8.5), ylim = c(0.8, 3.2)) +
theme_dag()
ggplot(data = DAG.ex4.3, aes(x = x, y = y, xend = xend, yend = yend)) +
#visualize causal effect path
geom_segment(x = 1, xend = 8.2, y = 3, yend = 3,
linewidth = 8, lineend = 'square', colour = '#D55E00', alpha = 0.05, linetype = '12') +
#latent variable
geom_point(x = 8, y = 3, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 8, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 3.5, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 1, y = 2, shape = 1, size = 18, stroke = 0.9, color = 'black') +
#adjusted variable
geom_point(x = 6, y = 1, shape = 22, size = 14, stroke = 0.9, fill = 'grey80', colour = 'black') +
geom_dag_text(aes(label = node_labels[name]),
parse = TRUE,
colour = 'black',
size = 10,
family = 'mono') +
geom_dag_edges(arrow_directed = grid::arrow(length = grid::unit(10, 'pt'), type = 'open'),
edge_colour = 'black',
family = 'mono',
fontface = 'bold') +
coord_cartesian(xlim = c(0.5, 8.5), ylim = c(0.8, 3.2)) +
theme_dag()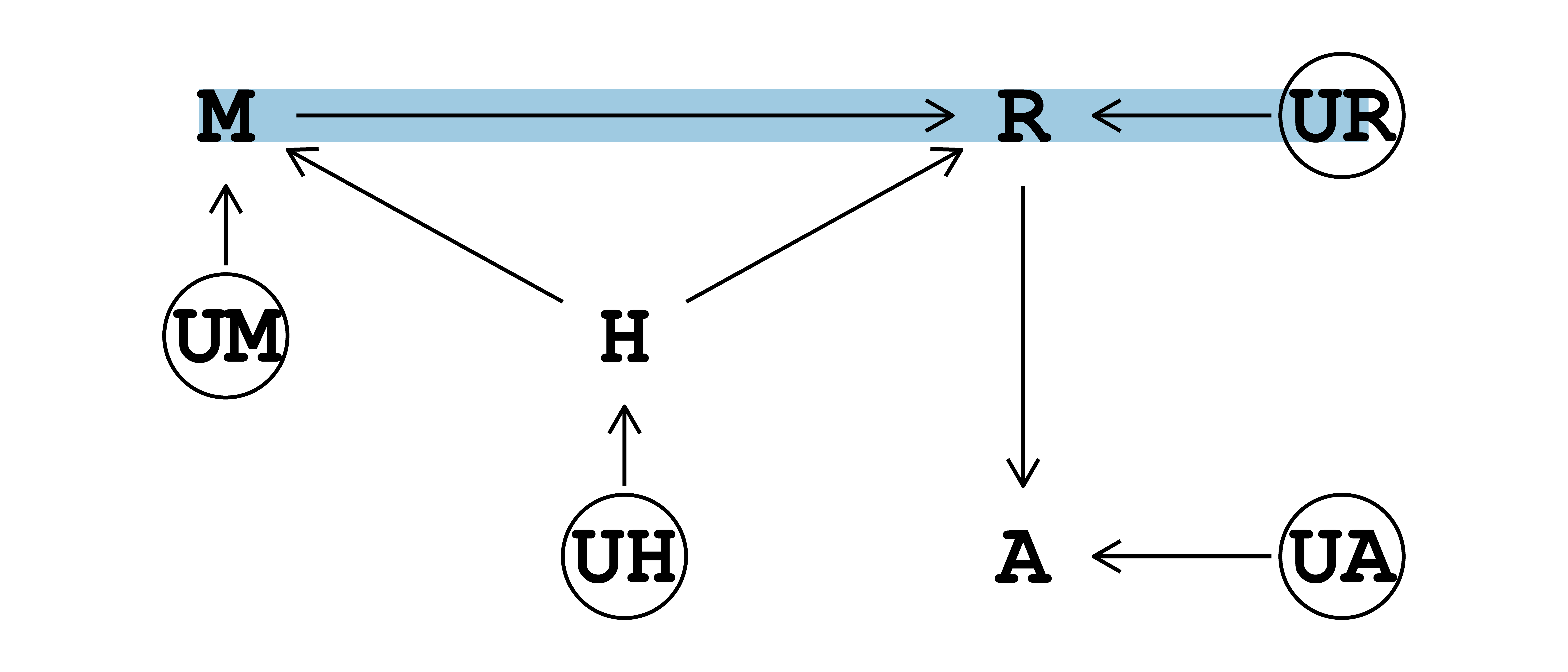
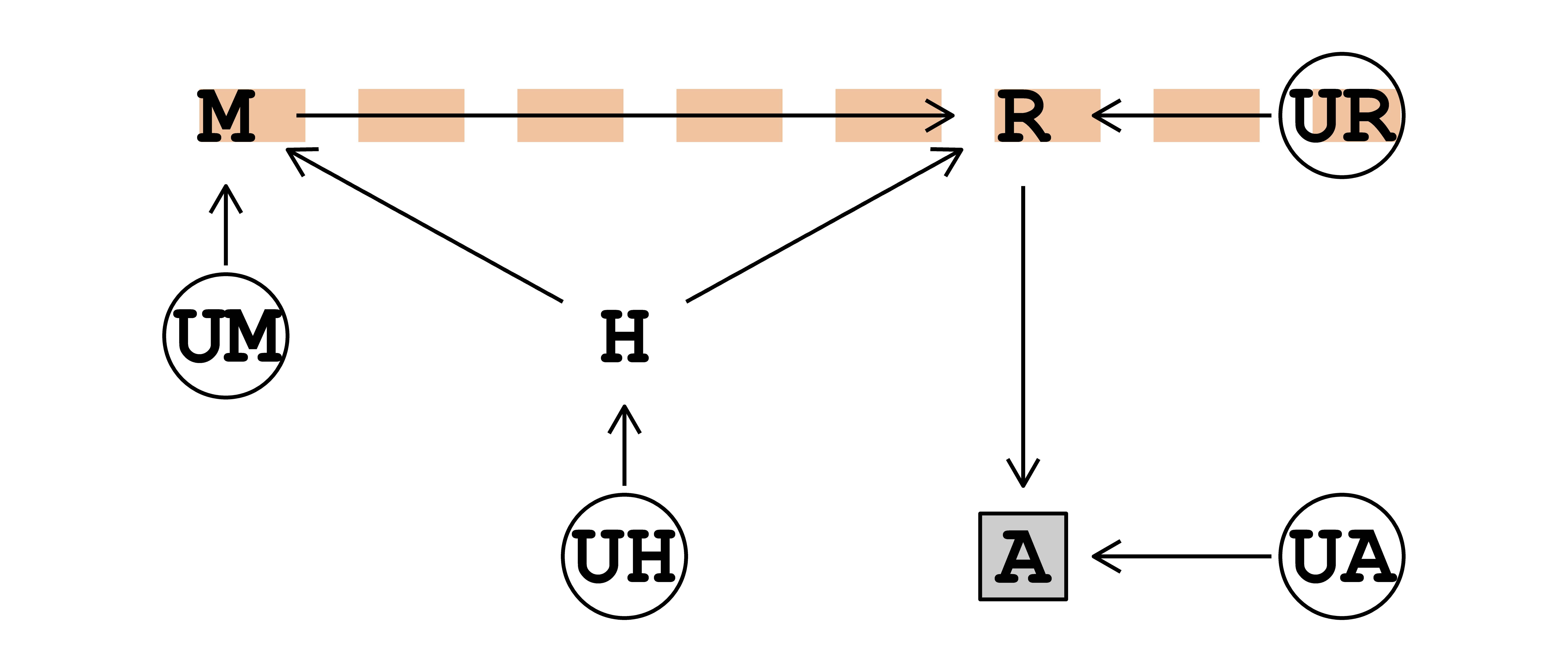
Fig. 4.6 shows the magnified DAG with the causes for all variables. Here the causes only affect one variable (i.e., they are not shared) and are circled because they are considered unobserved. Consequently, they are generally omitted in a DAG. This structure implies that, for example, respiratory irritation \((\text{R})\) is a function of microbial aerosol \((\text{M})\), relative humidity \((\text{H})\), and some unobserved influence \((\text{u}_\text{R})\). This unobserved influence (or set of influences) can be thought of as variations around the expected value of \(\text{R}\) for each \(\text{M}\) and \(\text{H}\).
From Fig. 4.6, it is clear that respiratory irritation \((\text{R})\) is a virtual collider on the path \(\text{M} \rightarrow \text{R} \leftarrow \text{u}_\text{R}\). This path is closed (Fig. 4.6 (a)), but conditioning on absence \(\text{A}\) partially opens it (Fig. 4.6 (b)). This happens because \(\text{A}\) is a descendant of \(\text{R}\), and adjusting for \(\text{A}\) is similar to adjusting for \(\text{R}\). Consequently, the non-causal path \(\text{M} \rightarrow \text{R} \leftarrow \text{u}_\text{R}\) is now partially open, creating a spurious association between \(\text{M}\) and \(\text{u}_\text{R}\) that distorts the estimate of the causal effect of \(\text{M}\) on \(\text{R}\). This is why the estimates from Model.1 are biased. Following the backdoor criterion, we should not adjust for any descendant of the cause of interest. Since Model.2 does not include absence, which is a descendant of microbial aerosol (i.e., \(\text{M} \rightarrow \text{R} \rightarrow \text{A}\)), the estimates from this model are unbiased.
Bayesian framework
#Fit the linear regression model with M, H, A and R (Model 1)
ex4_Model.1 <- stan_glm(formula = R ~ M + H + A,
family = gaussian(),
data = ex4_sample.random,
#Prior coefficients
prior = normal(location = c(0, 0, 0), scale = c(6, 1, 4)),
#Prior intercept
prior_intercept = normal(location = 0, scale = 2),
#Prior sigma
prior_aux = exponential(rate = 0.5),
iter = 4000, warmup = 1000,
save_warmup = TRUE,
chains = 4, cores = 4,
seed = 2025) #for reproducibility
#View of the model
ex4_Model.1stan_glm
family: gaussian [identity]
formula: R ~ M + H + A
observations: 5000
predictors: 4
------
Median MAD_SD
(Intercept) -2.9 0.1
Mhigh 1.4 0.1
Hlow 0.3 0.1
Ayes 4.6 0.1
Auxiliary parameter(s):
Median MAD_SD
sigma 2.0 0.0
------
* For help interpreting the printed output see ?print.stanreg
* For info on the priors used see ?prior_summary.stanreg#Fit the linear regression model with M, H and R (Model 2)
ex4_Model.2 <- stan_glm(formula = R ~ M + H,
family = gaussian(),
data = ex4_sample.random,
#Prior coefficients
prior = normal(location = c(0, 0), scale = c(6, 1)),
#Prior intercept
prior_intercept = normal(location = 0, scale = 2),
#Prior sigma
prior_aux = exponential(rate = 0.5),
iter = 4000, warmup = 1000,
save_warmup = TRUE,
chains = 4, cores = 4,
seed = 2025) #for reproducibility
#View of the model
ex4_Model.2stan_glm
family: gaussian [identity]
formula: R ~ M + H
observations: 5000
predictors: 3
------
Median MAD_SD
(Intercept) -1.0 0.1
Mhigh 2.9 0.1
Hlow 0.5 0.1
Auxiliary parameter(s):
Median MAD_SD
sigma 2.9 0.0
------
* For help interpreting the printed output see ?print.stanreg
* For info on the priors used see ?prior_summary.stanregIn Bayesian analysis, there are important diagnostics that have to be carried out in order to assess the convergence and efficiency of the Markov Chains. This is done by using the monitor() function which computes summaries of MCMC (Markov Chain Monte Carlo) draws and monitor convergence. Specifically, we will look at Rhat, Bulk_ESS and Tail_ESS metrics.
#Diagnostics for model 1
monitor(ex4_Model.1$stanfit) Inference for the input samples (4 chains: each with iter = 4000; warmup = 0):
Q5 Q50 Q95 Mean SD Rhat Bulk_ESS Tail_ESS
(Intercept) -3.1 -2.9 -2.8 -2.9 0.1 1 8175 7548
Mhigh 1.2 1.4 1.5 1.4 0.1 1 7316 7632
Hlow 0.2 0.3 0.5 0.3 0.1 1 7483 8012
Ayes 4.5 4.6 4.7 4.6 0.1 1 11050 7932
sigma 2.0 2.0 2.0 2.0 0.0 1 12676 9141
mean_PPD 0.7 0.8 0.9 0.8 0.0 1 12106 11301
log-posterior -10602.1 -10598.7 -10597.1 -10599.0 1.6 1 5391 7209
For each parameter, Bulk_ESS and Tail_ESS are crude measures of
effective sample size for bulk and tail quantities respectively (an ESS > 100
per chain is considered good), and Rhat is the potential scale reduction
factor on rank normalized split chains (at convergence, Rhat <= 1.05).#Diagnostics for model 2
monitor(ex4_Model.2$stanfit) Inference for the input samples (4 chains: each with iter = 4000; warmup = 0):
Q5 Q50 Q95 Mean SD Rhat Bulk_ESS Tail_ESS
(Intercept) -1.2 -1.0 -0.8 -1.0 0.1 1 6384 7117
Mhigh 2.7 2.9 3.1 2.9 0.1 1 6823 7134
Hlow 0.3 0.5 0.7 0.5 0.1 1 7009 7386
sigma 2.9 2.9 3.0 2.9 0.0 1 9718 8191
mean_PPD 0.7 0.8 0.9 0.8 0.1 1 10639 10331
log-posterior -12501.9 -12498.9 -12497.6 -12499.2 1.4 1 5257 6747
For each parameter, Bulk_ESS and Tail_ESS are crude measures of
effective sample size for bulk and tail quantities respectively (an ESS > 100
per chain is considered good), and Rhat is the potential scale reduction
factor on rank normalized split chains (at convergence, Rhat <= 1.05).Rhat is a metric used to assess the convergence of Markov Chain Monte Carlo (MCMC) simulations. It helps determine if the MCMC chains have adequately explored the target posterior distribution. Specifically, it compares the between- and within-chain estimates for model parameters: If chains have not mixed well (i.e., the between- and within-chain estimates do not agree), R-hat is larger than 1. A general rule of thumb is to use the sample only if R-hat is less than 1.05; a larger value suggests that the chains have not mixed well, and the results might not be reliable. In our two models, all Rhat are equal to 1 indicating that the chains have mixed well and have adequately explored the target posterior distribution.
Bulk_ESS and Tail_ESS stand for ‘Bulk Effective Sample Size’ and ‘Tail Effective Sample Size,’ respectively. Since MCMC samples are not truly independent (they are correlated), these metrics assess the sampling efficiencies, that is, they help evaluate how efficiently the MCMC sampler is exploring the parameter space.
-
Bulk_ESSis a useful measure for sampling efficiency in the bulk (center) of the distribution (e.g., efficiency of mean and median estimates); -
Tail_ESSis a useful measure for sampling efficiency in the tails of the distribution (e.g., efficiency of variance and tail quantile estimates). A general rule of thumb is that bothBulk-ESSandTail-ESSshould be at least 100 (approximately) per Markov Chain in order to be reliable and indicate that estimates of respective posterior quantiles are reliable. In our two models, allBulk-ESSandTail-ESSare well above 400 (i.e., 100 multiplied by 4, the number of chains we used) indicating that estimates of posterior quantiles are reliable.
Since we have established that the posteriors are reliable, we can now explore the model estimates. The estimated coefficients for the two models are then plotted in Fig. 4.7.
Code
#Extract draws from model 1
post_ex4_Model.1 <-
ex4_Model.1 %>%
spread_draws(Mhigh) %>% #extract draws from the fitted model
mutate(model = 'Model.1') #add a new column to specify that the model
#Extract draws from model 2
post_ex4_Model.2 <-
ex4_Model.2 %>%
spread_draws(Mhigh) %>% #extract draws from the fitted model
mutate(model = 'Model.2') #add a new column to specify that the model
#Combine draws
plot.post <- rbind(post_ex4_Model.1, post_ex4_Model.2)
# Plot
plot.post %>%
ggplot(aes(y = model, x = Mhigh)) +
stat_slabinterval(point_interval = 'mean_hdi',
.width = c(.95)) +
geom_vline(xintercept = b_micr.resp, alpha = 0.8, linetype = 'dashed', colour = 'blue') +
scale_x_continuous('Estimate',
breaks = seq(from = -5, to = 5, by = 0.5),
limits = c(0.5, 3.5)) +
theme(panel.grid = element_blank(),
panel.background = element_blank(),
panel.border = element_rect(colour = 'black', fill = NA),
axis.title.y = element_blank())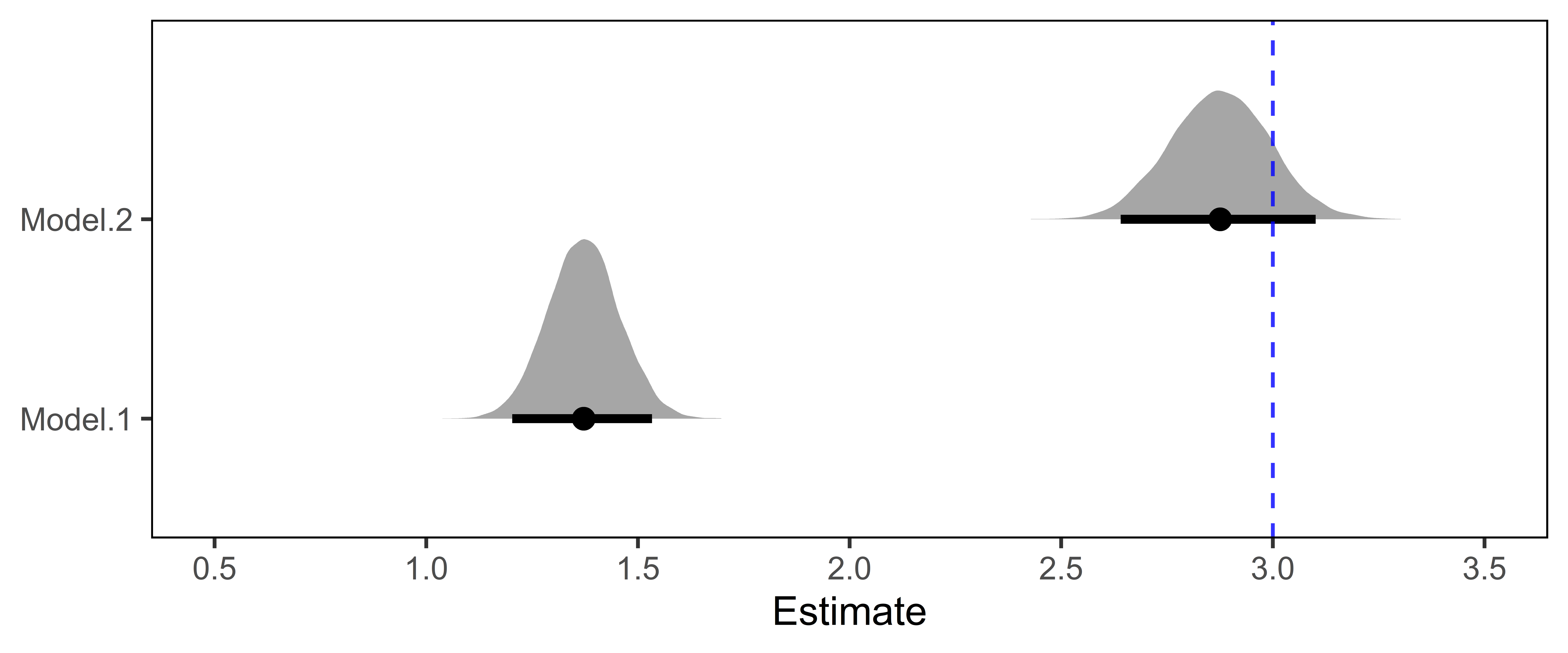
Fig. 4.7 shows the estimates (mean and 95% HDI) of the coefficient for microbial aerosol for Model.1, and Model.2.
For Model.1 we found a positive coefficient between microbial aerosol \((\text{M})\) and respiratory irritation \((\text{R})\) (mean = 1.372, 95% HDI [1.203, 1.533]). The estimated causal effect is biased, leading to the wrong conclusion that changing microbial aerosol from low to high causes an increase in lung infection by 1.372 units, on average.
For Model.2 we found a positive coefficient between microbial aerosol \((\text{M})\) and respiratory irritation \((\text{R})\) (mean = 2.875, 95% HDI [2.640, 3.101]). The estimated causal effect is unbiased, leading to the correct conclusion that changing microbial aerosol from low to high causes an increase in lung infection by 2.875 units, on average.
Importantly, for both Model.1 and Model.1, the 95% HDI is the range of parameter values within which the most credible 95% of the posterior distribution falls. Unlike a frequentist confidence interval, the Bayesian 95% HDI has a direct probabilistic meaning: every point inside the HDI has a higher probability density than any point outside the interval. Therefore, given the model, the prior and the data, we can say that there is a 95% probability that the data-generating parameter (i.e., b_micr.resp = 3) lies within the HDI. However, since Model.1 leads to a biased estimate, we will reach the wrong conclusion by stating that there is a 95% probability that the data-generating parameter lies within the [1.203, 1.533] interval. This probability is 0%.
As expected, adjusting for relative humidity \((\text{H})\) (i.e., using Model.2) leads to the correct estimate of the total average causal effect of microbial aerosol \((\text{M})\) on respiratory irritation \((\text{R})\). However, when adjusting for both relative humidity \((\text{H})\) and absence \((\text{A})\) (i.e., using Model.1) the estimates are biased. This happened because absence \((\text{A})\) is a descendant of respiratory irritation \((\text{R})\), which is a collider. Adjusting for a descendant of a collider is akin to adjusting for the collider itself, and, therefore, can introduce a similar bias.
To better explain this bias, we can plot the magnified DAG that shows that causes of all variables.
Code
dag_coords.ex4.3 <-
data.frame(name = c('M', 'H', 'A', 'R', 'UA', 'UH', 'UM', 'UR'),
x = c(1, 3.5, 6, 6, 8, 3.5, 1, 8),
y = c(3, 2, 1, 3, 1, 1, 2, 3))
DAG.ex4.3 <-
dagify(M ~ H + UM,
H ~ UH,
R ~ H + M + UR,
A ~ R + UA,
coords = dag_coords.ex4.3)
node_labels <- c(
M = 'bold(M)',
H = 'bold(H)',
A = 'bold(A)',
R = 'bold(R)',
UA = 'bold(UA)',
UH = 'bold(UH)',
UM = 'bold(UM)',
UR = 'bold(UR)'
)
ggplot(data = DAG.ex4.3, aes(x = x, y = y, xend = xend, yend = yend)) +
#visualize causal effect path
geom_segment(x = 1, xend = 8, y = 3, yend = 3,
linewidth = 8, lineend = 'square', colour = '#0072B2', alpha = 0.05, linetype = 'solid') +
geom_point(x = 8, y = 3, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 8, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 3.5, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 1, y = 2, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_dag_text(aes(label = node_labels[name]),
parse = TRUE,
colour = 'black',
size = 10,
family = 'mono') +
geom_dag_edges(arrow_directed = grid::arrow(length = grid::unit(10, 'pt'), type = 'open'),
edge_colour = 'black',
family = 'mono',
fontface = 'bold') +
coord_cartesian(xlim = c(0.5, 8.5), ylim = c(0.8, 3.2)) +
theme_dag()
ggplot(data = DAG.ex4.3, aes(x = x, y = y, xend = xend, yend = yend)) +
#visualize causal effect path
geom_segment(x = 1, xend = 8.2, y = 3, yend = 3,
linewidth = 8, lineend = 'square', colour = '#D55E00', alpha = 0.05, linetype = '12') +
#latent variable
geom_point(x = 8, y = 3, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 8, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 3.5, y = 1, shape = 1, size = 18, stroke = 0.9, color = 'black') +
geom_point(x = 1, y = 2, shape = 1, size = 18, stroke = 0.9, color = 'black') +
#adjusted variable
geom_point(x = 6, y = 1, shape = 22, size = 14, stroke = 0.9, fill = 'grey80', colour = 'black') +
geom_dag_text(aes(label = node_labels[name]),
parse = TRUE,
colour = 'black',
size = 10,
family = 'mono') +
geom_dag_edges(arrow_directed = grid::arrow(length = grid::unit(10, 'pt'), type = 'open'),
edge_colour = 'black',
family = 'mono',
fontface = 'bold') +
coord_cartesian(xlim = c(0.5, 8.5), ylim = c(0.8, 3.2)) +
theme_dag()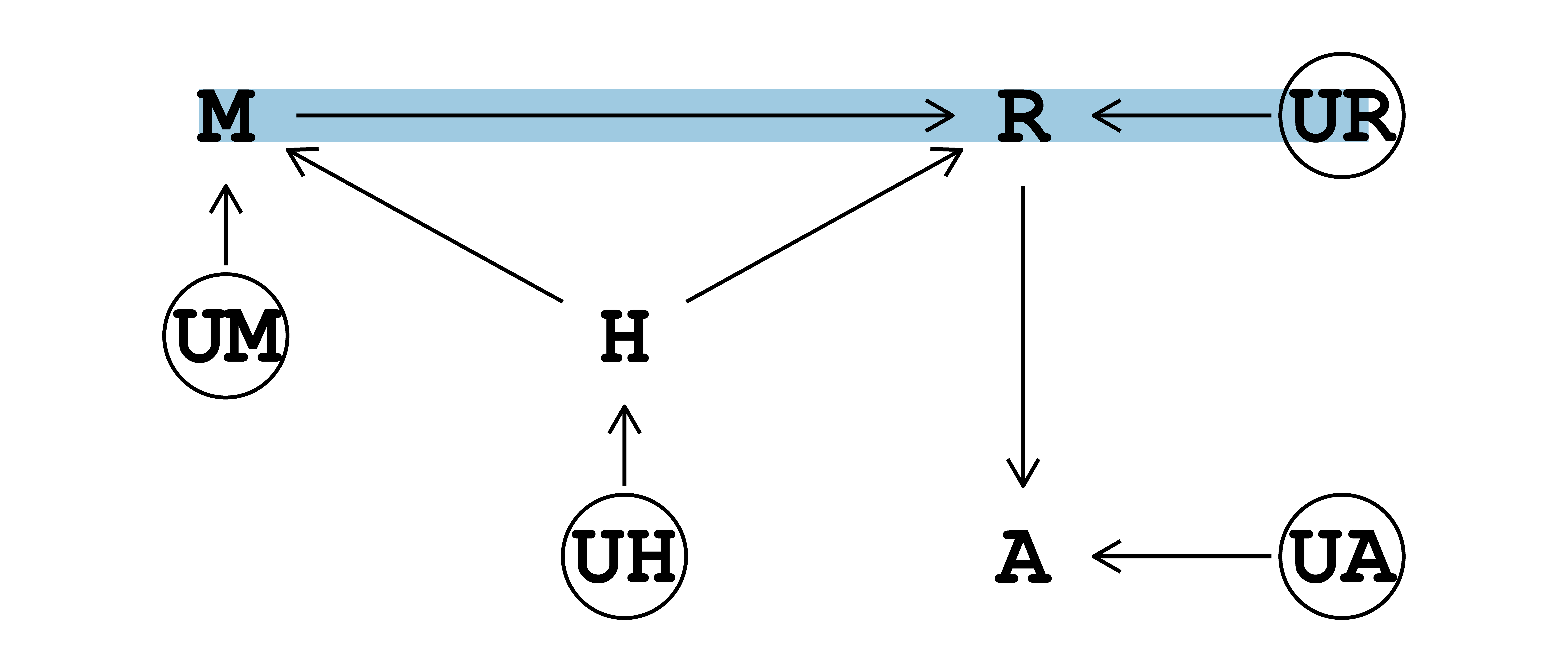
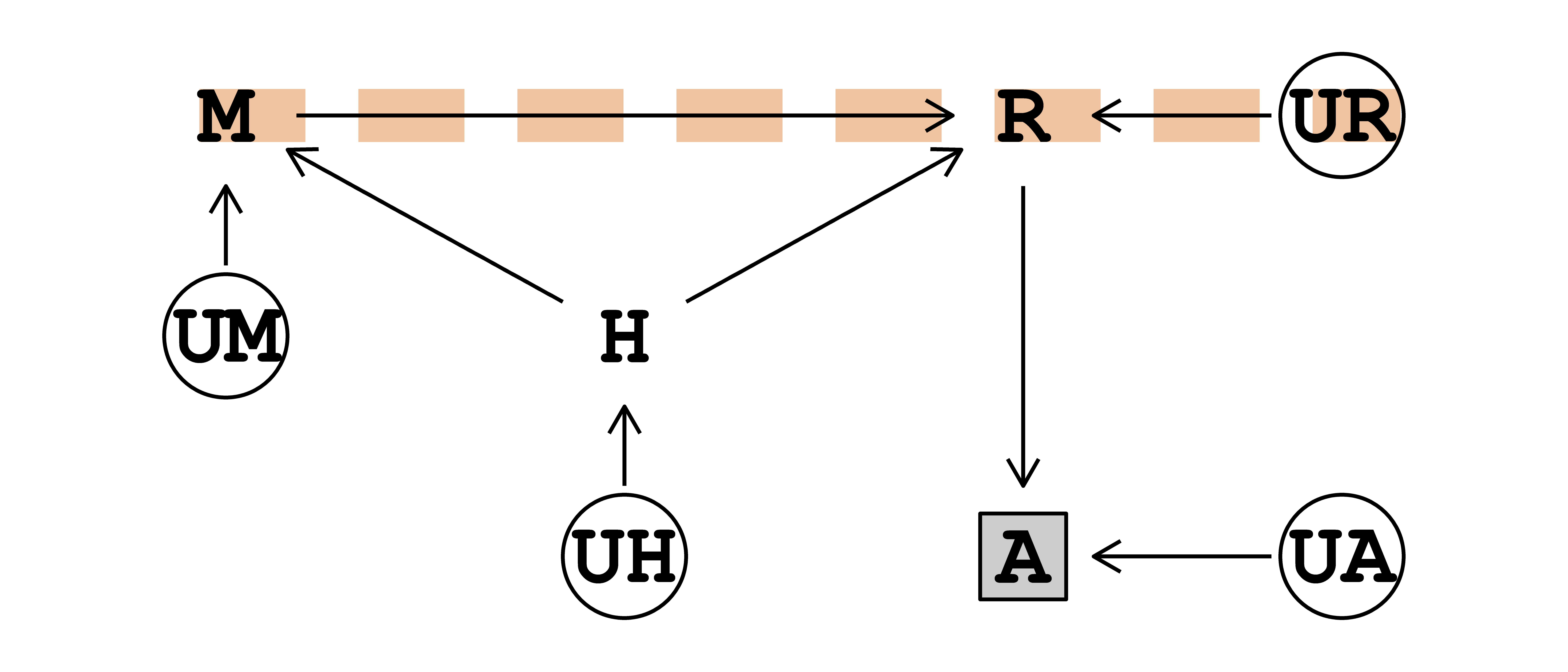
Fig. 4.8 shows the magnified DAG with the causes for all variables. Here the causes only affect one variable (i.e., they are not shared) and are circled because they are considered unobserved. Consequently, they are generally omitted in a DAG. This structure implies that, for example, respiratory irritation \((\text{R})\) is a function of microbial aerosol \((\text{M})\), relative humidity \((\text{H})\), and some unobserved influence \((\text{u}_\text{R})\). This unobserved influence (or set of influences) can be thought of as variations around the expected value of \(\text{R}\) for each \(\text{M}\) and \(\text{H}\).
From Fig. 4.6, it is clear that respiratory irritation \((\text{R})\) is a virtual collider on the path \(\text{M} \rightarrow \text{R} \leftarrow \text{u}_\text{R}\). This path is closed (Fig. 4.8 (a)), but conditioning on absence \(\text{A}\) partially opens it (Fig. 4.8 (b)). This happens because \(\text{A}\) is a descendant of \(\text{R}\), and adjusting for \(\text{A}\) is similar to adjusting for \(\text{R}\). Consequently, the non-causal path \(\text{M} \rightarrow \text{R} \leftarrow \text{u}_\text{R}\) is now partially open, creating a spurious association between \(\text{M}\) and \(\text{u}_\text{R}\) that distorts the estimate of the causal effect of \(\text{M}\) on \(\text{R}\). This is why the estimates from Model.1 are biased. Following the backdoor criterion, we should not adjust for any descendant of the cause of interest. Since Model.2 does not include absence, which is a descendant of microbial aerosol (i.e., \(\text{M} \rightarrow \text{R} \rightarrow \text{A}\)), the estimates from this model are unbiased.
Session info
Version information about R, the OS and attached or loaded packages.
R version 4.2.3 (2023-03-15 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 26200)
Matrix products: default
locale:
[1] LC_COLLATE=English_United Kingdom.utf8
[2] LC_CTYPE=English_United Kingdom.utf8
[3] LC_MONETARY=English_United Kingdom.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United Kingdom.utf8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] rstan_2.32.7 StanHeaders_2.32.10 gt_1.1.0
[4] lubridate_1.9.4 forcats_1.0.1 stringr_1.6.0
[7] dplyr_1.1.4 purrr_1.2.0 readr_2.1.6
[10] tidyr_1.3.1 tibble_3.3.0 ggplot2_4.0.1
[13] tidyverse_2.0.0 ggdist_3.3.3 tidybayes_3.0.7
[16] rstanarm_2.32.2 Rcpp_1.1.0 dagitty_0.3-4
[19] ggdag_0.2.13
loaded via a namespace (and not attached):
[1] backports_1.5.0 plyr_1.8.9 igraph_2.2.1
[4] splines_4.2.3 svUnit_1.0.8 crosstalk_1.2.2
[7] rstantools_2.5.0 inline_0.3.21 digest_0.6.38
[10] htmltools_0.5.8.1 viridis_0.6.5 magrittr_2.0.4
[13] checkmate_2.3.3 memoise_2.0.1 tzdb_0.5.0
[16] graphlayouts_1.2.2 RcppParallel_5.1.11-1 matrixStats_1.5.0
[19] xts_0.14.1 timechange_0.3.0 colorspace_2.1-2
[22] ggrepel_0.9.6 rbibutils_2.4 xfun_0.54
[25] jsonlite_2.0.0 lme4_1.1-37 survival_3.8-3
[28] zoo_1.8-14 glue_1.8.0 reformulas_0.4.2
[31] polyclip_1.10-7 gtable_0.3.6 V8_8.0.1
[34] distributional_0.5.0 car_3.1-3 pkgbuild_1.4.8
[37] abind_1.4-8 scales_1.4.0 miniUI_0.1.2
[40] viridisLite_0.4.2 xtable_1.8-4 Formula_1.2-5
[43] stats4_4.2.3 DT_0.34.0 htmlwidgets_1.6.4
[46] threejs_0.3.4 arrayhelpers_1.1-0 RColorBrewer_1.1-3
[49] posterior_1.6.1 pkgconfig_2.0.3 loo_2.8.0
[52] farver_2.1.2 sass_0.4.10 utf8_1.2.6
[55] tidyselect_1.2.1 labeling_0.4.3 rlang_1.1.6
[58] reshape2_1.4.5 later_1.4.4 tools_4.2.3
[61] cachem_1.1.0 cli_3.6.5 generics_0.1.4
[64] evaluate_1.0.5 fastmap_1.2.0 yaml_2.3.10
[67] knitr_1.50 fs_1.6.6 tidygraph_1.3.1
[70] ggraph_2.2.2 nlme_3.1-168 mime_0.13
[73] xml2_1.5.0 compiler_4.2.3 bayesplot_1.14.0
[76] shinythemes_1.2.0 rstudioapi_0.17.1 curl_7.0.0
[79] tweenr_2.0.3 stringi_1.8.7 lattice_0.22-7
[82] Matrix_1.6-5 nloptr_2.2.1 markdown_2.0
[85] shinyjs_2.1.0 tensorA_0.36.2.1 vctrs_0.6.5
[88] pillar_1.11.1 lifecycle_1.0.4 Rdpack_2.6.4
[91] httpuv_1.6.16 QuickJSR_1.8.1 R6_2.6.1
[94] promises_1.5.0 gridExtra_2.3 codetools_0.2-20
[97] boot_1.3-32 colourpicker_1.3.0 MASS_7.3-58.2
[100] gtools_3.9.5 withr_3.0.2 shinystan_2.6.0
[103] parallel_4.2.3 hms_1.1.4 grid_4.2.3
[106] coda_0.19-4.1 minqa_1.2.8 rmarkdown_2.30
[109] S7_0.2.1 carData_3.0-5 otel_0.2.0
[112] ggforce_0.5.0 shiny_1.11.1 base64enc_0.1-3
[115] dygraphs_1.1.1.6